DCDA is a "Discrete Charge Dipole Approximation" program that enables the computation
of static and frequency-dependent polarizabilities. The program relies
on an electrostatic model in which the atoms of the structures considered are
represented by a net electric charge and by a dipole. The net electric charge
completes the representation found in DDA (Discrete Dipole Approximation) models,
which only account for discrete dipoles. This model accounts for the displacement
of free charges, for the accumulation of additional charges
and for the transfer of charges between the structures considered and a metallic
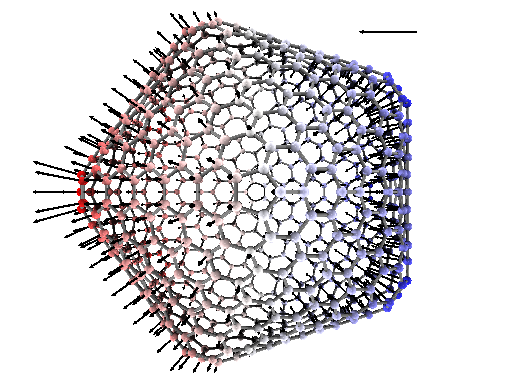
substrate. In addition to the atomic charges and dipoles, the program provides
the permanent polarization of the structures considered, their molecular polarizability
as well as local electric fields that keep meaningful down to the atomic level.
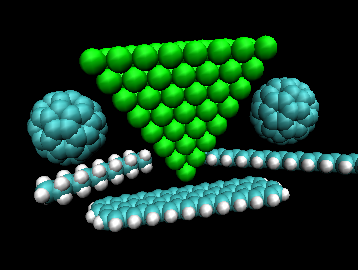
The model has been parametrized so far for carbon nanotubes, fullerenes, alkanes,
alkenes, aromatic molecules and silver clusters. The program makes it possible
to consider different structures in the same simulation, each structure being described by
its own set of parameters and having its own charge-neutrality condition implemented.
The program is written in Fortran 90 in a user-friendly style, which enables the program to
be easily applied to a wide range of problems.
The DCDA software is an implementation of the theory developed in the following articles :
If you are interested by this software, please contact the author.